We co-exist with zillions of other living organisms on our planet earth. From the tiny single-celled bacteria to the giant and mammoth blue whale, every living being has traced its path and has a tale to tell of how they survived on this planet. Varied they may seem, every living being is made up of cells, and what makes all the difference is the DNA which resides inside these cells. Even before its discovery in the 1950s to this date, deciphering the language of DNA has remained an interesting subject for researchers. Armed with the high throughput machines that sequence DNA in no time, researchers across the globe are now trying to understand the changes in DNA that have led to a variety of life forms.
One interesting area of research in this field is predicting the essential genes, which are found in various organisms, as they are required to carry out common biological functions. Knowledge of such common genes in microbes can help understand microbial growth and survival, and also in making broad-spectrum antibiotics to treat infections. However, one major issue that currently limits research in this area is the difficulty in predicting essential genes. Given the experimental identification of essential genes across DNA is an arduous task, scientists are trying different prediction methods to find essential genes across various genomes. Once predicted through relatively inexpensive and quick computational methods, the function of the predicted gene can be verified by fewer targeted laboratory experiments.
Dr Karthik Raman, who is an Associate Professor at IIT Madras and the Robert Bosch Center of Data Science and Artificial Intelligence, has been working on this issue for quite some time now. In their recent research, which has been published in the international journal PLoS ONE, Dr Raman along with his colleagues has reported the discovery of gene features that enable prediction of essential genes across diverse organisms. To ensure that the information obtained can be used by the research community, the team has also made a database named NetGenes.
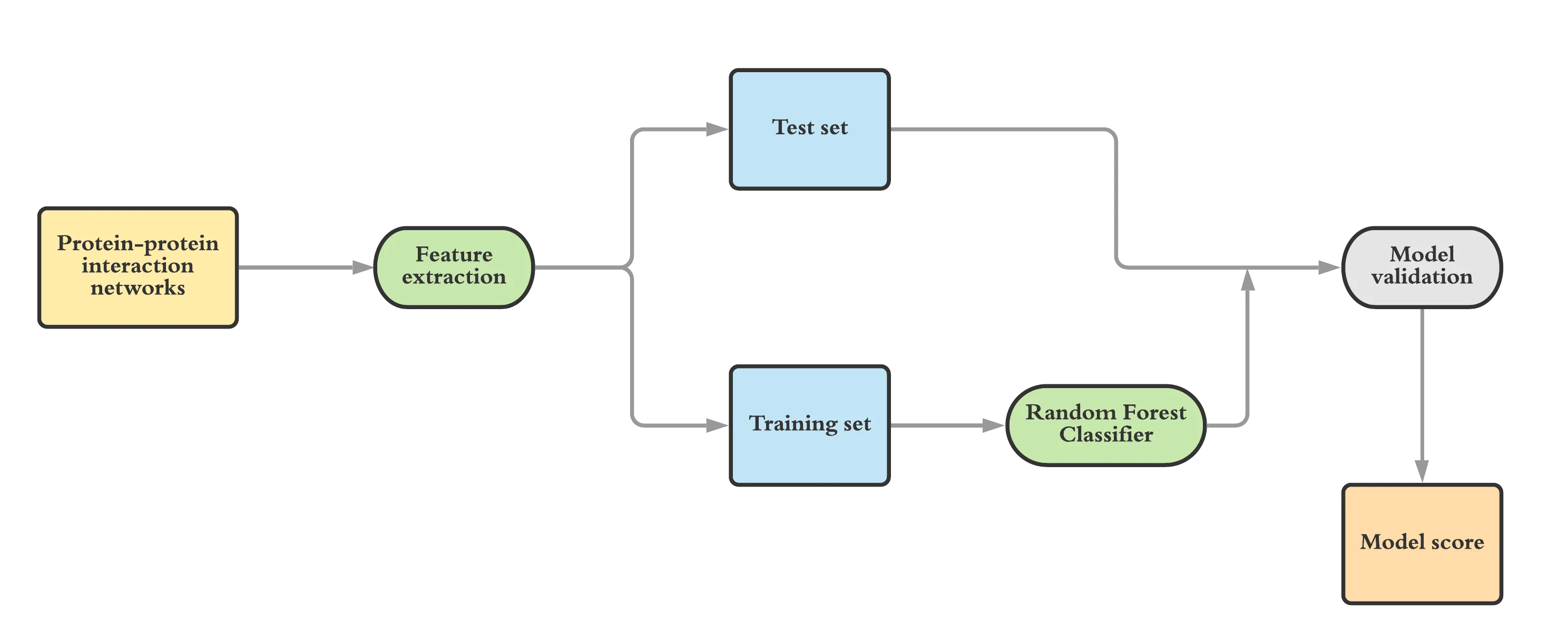
Figure showing workflow of the machine learning model
“A classic challenge in biology is to study the function of proteins. Of various functions, essential functions are very interesting, as they map to important indispensable genes in an organism. Experimentally identifying these genes is rather expensive and challenging. Computational predictions can help point in the right direction, to prioritise experiments. To date, experimental data are available for <100 organisms! On the other hand, sequencing data are available for 1000s of organisms, as also interactome (networks of interactions) data. In the paper, we posit that network information is crucial to predict essentiality and achieve excellent performance in prediction, by simply using network data,” explains Dr. Raman.
While various other approaches have been used in the past for the discovery of genes, they are narrow in their approach limiting their applicability. In this study, scientists studied 31 microbial genomes and used machine learning to derive features which are defining of essential genes in these 31 microbial genomes. Many genes work together to carry out a biological process making them a part of the biological network. Scientists utilize this information in the search of essential genes. The idea was to have these network features and then utilize them to identify genes from the organisms where genomes have been sequenced but the gene essentiality information is missing.
“Conventional methods rely too much on what is known, and use sequence/protein-based information (from existing organisms that are well studied), to make predictions on essentiality in other organisms. Surprisingly, very few studies have used the power of very large, network-based predictions, to find features of genes that might be essential or not. The power of this system is that it minimizes bias considerably, and does not make too many assumptions. Instead, using given genome data, it makes predictions on properties of the network that is being studied. This study makes excellent use of network-based properties to predict if a gene will be essential or not, and in fact is a substantial improvement over sequence-based predictions. As information in databases improve, this method is very likely to become even more powerful in predicting essential genes,” comments Dr. Sunil Laxman, Assistant Investigator at InStem and also a DBT- India Alliance Intermediate fellow.
For a wider application of this research, the team has hosted a program based on these features on the website of Robert Bosch Center of Artificial Intelligence and Data Science (https://rbc-dsai-iitm.github.io/NetGenes/). The program has been used to predict essential genes for 2711 bacterial organisms using network-based features derived in the research. The research community can utilize this information while studying the respective bacteria. The knowledge will have various biotechnological and industrial applications.
“The fact that we are able to make good predictions with network data enables us to leverage large amounts of interactome data available to make predictions, this is what NetGenes database now does, predicting putative essential genes at a massive scale,” adds Dr. Raman.
The team hasn’t stopped at this feat of theirs! They are further working to refine their method to further improve gene prediction.
Link to the article: https://journals.plos.org/plosone/article/comments?id=10.1371/journal.pone.0208722
Preprint: https://www.biorxiv.org/content/10.1101/2020.12.17.423287v1 (pre-print of the paper discussing the NetGenes database)
Keywords
Essential genes, database, network analytics, bacteria